Calculation of an IR Spectrum¶
This tutorial demonstrates the calculation of an IR spectrum using the ORCA python interface (OPI).
In this notebook we will:
Import the required Python dependencies
Define a working directory
Set up an input structure for our calculation
Perform a frequency calculation in the gas-phase
Parse the results and plot it
Step 1: Import Dependencies¶
We start by importing the modules needed for:
Interfacing with ORCA input/output
Numerical calculations and data handling
Plotting results
Note: We additionally import modules for visualization/plotting like
py3Dmol. For this, it might be necessary to installpy3Dmolinto your OPIvenv(e.g., by activating the.venvand usinguv pip install py3Dmol).
# > Import pathlib for directory handling
from pathlib import Path
import shutil
# > Import numpy for data handling and numerical operations
import numpy as np
# > OPI imports for performing ORCA calculations and reading output
from opi.core import Calculator
from opi.input.simple_keywords import Dft, Task, BasisSet, Approximation, AuxBasisSet, DispersionCorrection
from opi.input.structures.structure import Structure
from opi.input.blocks.block_freq import BlockFreq
# > Import libraries for visualization
import matplotlib.pyplot as plt
import py3Dmol
Step 2: Define Working Directory¶
All actual calculations will be performed in a subfolder RUN_09.
# > Calculation is performed in `RUN`
working_dir = Path("RUN")
# > The `working_dir`is automatically (re-)created
shutil.rmtree(working_dir, ignore_errors=True)
working_dir.mkdir()
Step 3: Setup an Input Structure¶
We use methanol as our example molecule. The 3D structure in Cartesian coordinates is defined in XYZ format and visualized.
# > Define the molecule's Cartesian coordinates in Angstroem as python string
xyz_data = """\
7
O 1.14430000000000 0.24120000000000 0.00000000000000
C -1.25740000000000 0.18150000000000 0.00000000000000
C 0.11300000000000 -0.42260000000000 0.00000000000000
H -1.79380000000000 -0.14930000000000 0.89240000000000
H -1.18650000000000 1.27190000000000 0.00160000000000
H -1.79280000000000 -0.14680000000000 -0.89380000000000
H 0.14780000000000 -1.52520000000000 -0.00070000000000\n
"""
# > Visualize the molecular structure using py3Dmol
view = py3Dmol.view(width=500, height=500)
view.addModel(xyz_data, "xyz")
view.setStyle({'stick': {'radius': 0.1}, 'sphere': {'scale': 0.2}})
view.setBackgroundColor('white')
view.zoomTo()
view.show()
# > Save the XYZ structure to a file
with open(working_dir / "struc.xyz","w") as f:
f.write(xyz_data)
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
Step 4: Calculation in the Gas-Phase¶
We run a geometry optimization and frequency calculation
# > Set up path and create directory for implicit model calculation
xyz_file = working_dir / "struc.xyz"
# > Create a Calculator object for ORCA input generation and execution
calc = Calculator(basename="gas", working_dir=working_dir)
# > Load the molecular structure from XYZ file
structure = Structure.from_xyz(xyz_file)
calc.structure = structure
calc.structure.charge = 0
calc.structure.multiplicity = 1
# > Add calculation keywords
calc.input.add_simple_keywords(
Dft.B3LYP, # > B3LYP method
BasisSet.AUG_CC_PVDZ, # > daug-cc-pVDZ basis set
AuxBasisSet.DEF2_J, # > auxiliary basis set
Approximation.RIJCOSX, # > Use RIJ and COSX
DispersionCorrection.D4, # > Use D4 model
Task.FREQ # > Do analytical Hessian
)
calc.input.add_blocks(
BlockFreq(scalfreq=0.970) # > Optimal scaling factor for B3LYP/aug-cc-pVDZ
)
# > Write the ORCA input file
calc.write_input()
# > Run the ORCA calculation
print("Running ORCA frequency calculation ...", end="")
calc.run()
print(" Done")
Running ORCA frequency calculation ... Done
Step 5: Parse Vibrational Spectrum and Plot it¶
We define a function for extracting the vibrational spectrum from the ORCA and plot it.
def plot_vibrational_spectrum(vibrational_spectrum: Path) -> None:
wave_numbers = []
intensities = []
# > While frequencies are available in OPI,
# > intensities are currently not.
# > Therefore, we grep them manually.
with open(vibrational_spectrum) as f:
in_block = False
for line in f:
if not in_block and "IR SPECTRUM" in line:
in_block = True
for _ in range(5):
next(f)
elif in_block and not line.strip(): # end of block
break
elif in_block:
parts = line.split()
try:
wave_number = float(parts[1])
intensity = float(parts[3])
if wave_number > 0 and intensity > 0:
wave_numbers.append(wave_number)
intensities.append(intensity)
except ValueError:
continue
# > Generate IR spectrum by creating a zero baseline and populating intensity values
max_wave = int(max(wave_numbers)) + 100
full_range = np.arange(0, max_wave, 1)
spectrum = np.zeros_like(full_range, dtype=float)
for wn, inten in zip(wave_numbers, intensities):
index = int(wn)
if index < len(spectrum):
spectrum[index] = inten
# > Plot the vibrational IR spectrum
plt.figure(figsize=(10, 5))
plt.plot(full_range, spectrum)
plt.xlabel('Wavenumber (cm⁻¹)')
plt.ylabel('IR Intensity (km/mol)')
plt.title('Vibrational Spectrum')
plt.grid(True)
plt.xlim(0, max_wave)
plt.ylim(0, max(intensities))
plt.tight_layout()
plt.show()
# > Plot vibrational spectrum for implicit solvation result
spectrum_path = working_dir / "gas.out"
plot_vibrational_spectrum(spectrum_path)
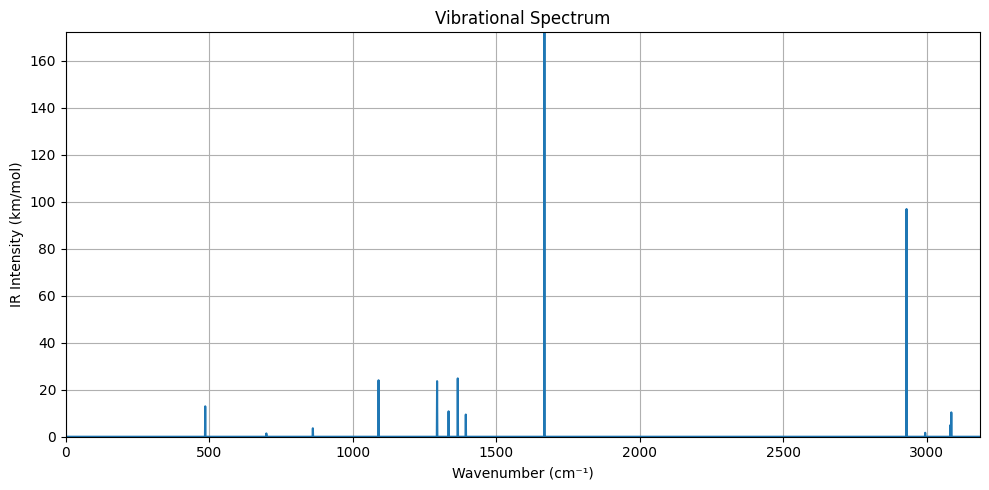
Summary¶
In this notebook we demonstrated how the ORCA Python interface can be employed to calculate the frequencies and intensities required for predicting an IR spectrum.